01 它能做什么
- 从口袋中结合的片段或配体出发做结构基础的方向性生长。
- 秒级筛选预计算的大型片段/构建块数据库。
- 单次运行最多输出 100,000 个生长结果。
- 可控制冲突、姿势优化、活性基团移除,易于嵌入自动化流程。
工作原理
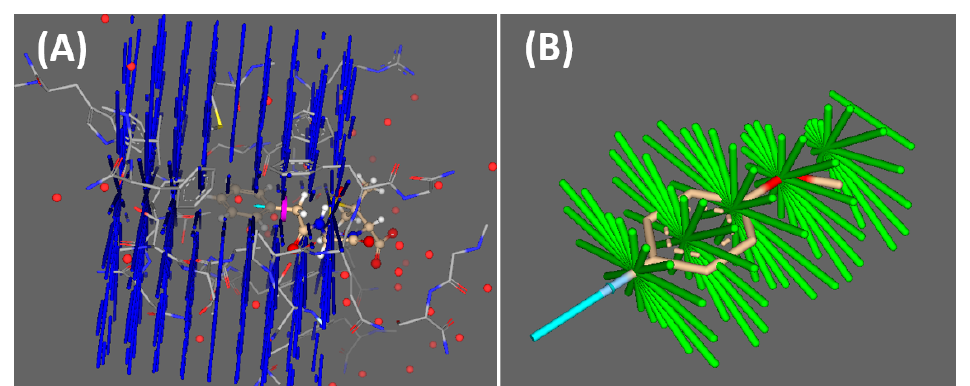
FastGrow 方法基于圆柱形形状描述符(ray volume matrices,RVM 描述符),沿生长方向描述口袋的空闲与被占据空间(Figure 1A);片段/构建块对应的形状描述符则预存于 FastGrow 数据库中(Figure 1B)。这些描述符为极快的形状匹配做了优化——惊人的速度来自用位移(bit-shifts)来模拟旋转。筛选时以口袋描述符为查询,与库中预计算的片段描述符匹配,按立体契合度打分排序;顶端片段被附着到核心上,姿势再经精修、并过滤掉残留冲突与附着时可能形成的活性基团,通过后过滤的化合物才报告给用户。在多个 self-growing 与 cross-growing 场景中,该形状描述符表现优异,在最多 84% 的案例中以小于 2 Å 的偏差重现了晶体结构构象。
02 前提与授权
需要 FastGrow 程序包、命令行环境与有效许可证。SeeSAR 许可证可用于 FastGrow,但不能用于 FastGrowDBCreator——后者需要单独的许可证。许可证放置方式同其他工具。
./fastgrow --license-info
03 Jump Start:一次片段生长
最少需要:一个片段数据库文件(.fastgrowdb,可从官网下载或用 FastGrowDBCreator 自建),加上位点+核心定义(两种方式)。
方式 A — 蛋白 + 核心配体
./fastgrow -d FragmentLibrary.fastgrowdb -p MyProtein.pdb \ -c MyCoreLigand.sdf -o MyGrowingOutput.sdf
方式 B — FastGrow 定义文件
从 SeeSAR 的 Inspirator Mode 导出 .fastgrow 文件,内含蛋白、结合位点、核心与 linker。
./fastgrow -d FragmentLibrary.fastgrowdb -f MyDefinitionFile.fastgrow \ -o MyGrowingOutput.sdf
默认输出最多 100 个结果,每个结果带打分字段 FASTGROW_OPTIMIZED_SCORE 或 FASTGROW_SCORE(取决于是否做了优化)。
04 程序选项
FastGrow 数据库(.fastgrowdb),含预处理好的片段/构建块。官网提供最多约 12 万化合物的现成库,也可自建。
蛋白文件,须配合 -c。用定义文件(-f)时此项作废。
核心分子(要从它生长)。须含 3D 坐标、位于口袋内,且至少有一个 linker 原子,该键定义 exit vector(生长方向)。
从 SeeSAR Inspirator Mode 导出的 .fastgrow 文件,含核心、exit vector、位点定义,可选药效团约束。与 -p 互斥。
结果文件(.sdf)。
最大结果数,允许 1–100,000。
当核心含多个 linker 时,指定要从哪个生长。格式为 R 加原子 ID,如 -l R1。
过滤/优化开关
| 选项 | 说明 |
|---|---|
--no-optimization | 关闭生长后的姿势优化(默认开启)。关闭后打分字段为 FASTGROW_SCORE。 |
--no-clash-filter | 关闭移除严重冲突姿势的过滤(默认开启)。 |
--no-smarts-filter | 关闭移除活性/不需要基团的 SMARTS 过滤(规则硬编码,仅含过氧基等高活性物种)。 |
05 准备 core 与 .fastgrowdb
准备 FastGrow 定义文件(推荐)
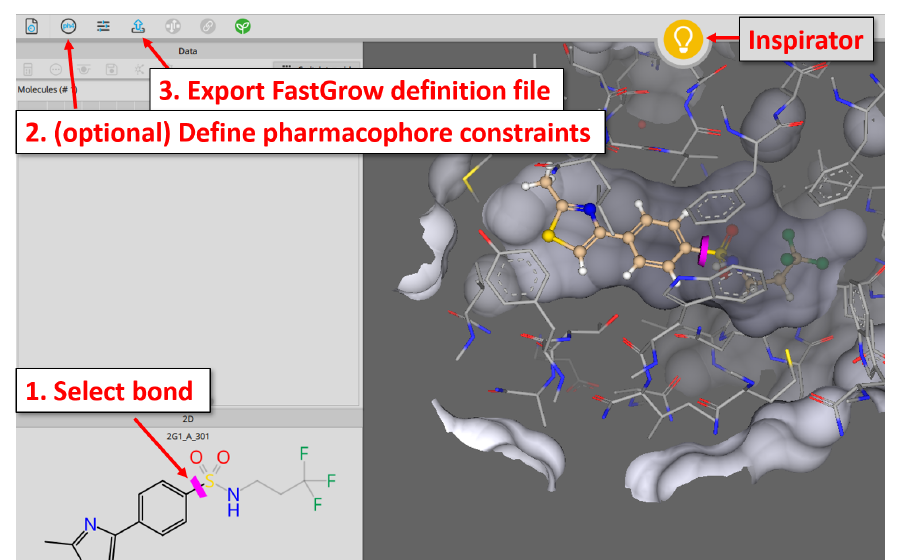
在命令行运行 FastGrow,推荐的方式是提供一个在 SeeSAR 里准备好的 .fastgrow 定义文件。在 SeeSAR 中下载一个共晶结构(如 PDB 4M3G)、提取配体后转入 Inspirator Mode,三步即可:① 在 2D 或 3D 窗口点击合适的键来定义 exit vector(要被替换的那部分会变灰;再次点击该键可反转生长方向);②(可选)用 ph4 按钮定义药效团约束,FastGrow 会据此过滤出符合约束的生长姿势;③ 导出定义文件,用命令行的 -f 指定即可。
准备核心配体(手动方式)
另一种方式是提供蛋白 + 一个含 linker 原子的独立核心配体文件(若已导出 .fastgrow 则无需此步)。以 4M3G 为例:在 SeeSAR 中提取配体 → 进 Molecule Editor Mode → 选中要删的原子、点垃圾桶图标删掉要被替换的部分 → 在剩余核心上选一个原子(本例为残留 SH 的硫原子),在 "Change element" 标签里选 [R] 把它改成 linker → 存到 Molecules Table 再导出为 SD 文件。然后命令行用 -c 指定它,并用 -p 指定对应蛋白(本例 4M3G.pdb)。可在单个核心里定义多个 linker 朝不同方向生长,但单次运行只能用一个 exit vector,用 -l 选(如 -l R1)。在 SeeSAR 中按住 L 键点击 linker 可查看其原子 ID;也可用文本编辑器打开 SD 文件,手动找标记为 R 或 R# 的 linker 原子 ID。
用 FastGrowDBCreator 自建数据库
要用自己的片段,需用 fastgrow_db_creator 预计算描述符。它需要单独许可证。每个输入片段必须恰好含一个 linker 原子。
./fastgrow_db_creator -i MyFragmentsWithLinker.sdf \ -o MyDatabase.fastgrowdb -n 100
| 选项 | 默认 | 说明 |
|---|---|---|
-i / --input | — | 片段(.smi .smiles .mol .mol2 .sdf),每个须恰好一个 linker,否则跳过。 |
-o / --output | — | 输出 .fastgrowdb。 |
-n / --nof-conformations | 10 | 每个片段生成的构象数上限。 |
-m / --mode | create | 执行模式:create 新建 / overwrite 覆盖 / append 追加到已有库(便于逐步建大库)。 |
--no-smarts-filter | — | 关闭活性基团过滤。 |
--error-output | — | 为未能入库的片段写出详细错误文件。 |